Sàng lọc rối loạn dự trữ lysosome (LSD) ở trẻ sơ sinh_Bệnh liên quan đến tích đọng các chất ở lysosome (Lysosomal Storage Diseases LSDs)
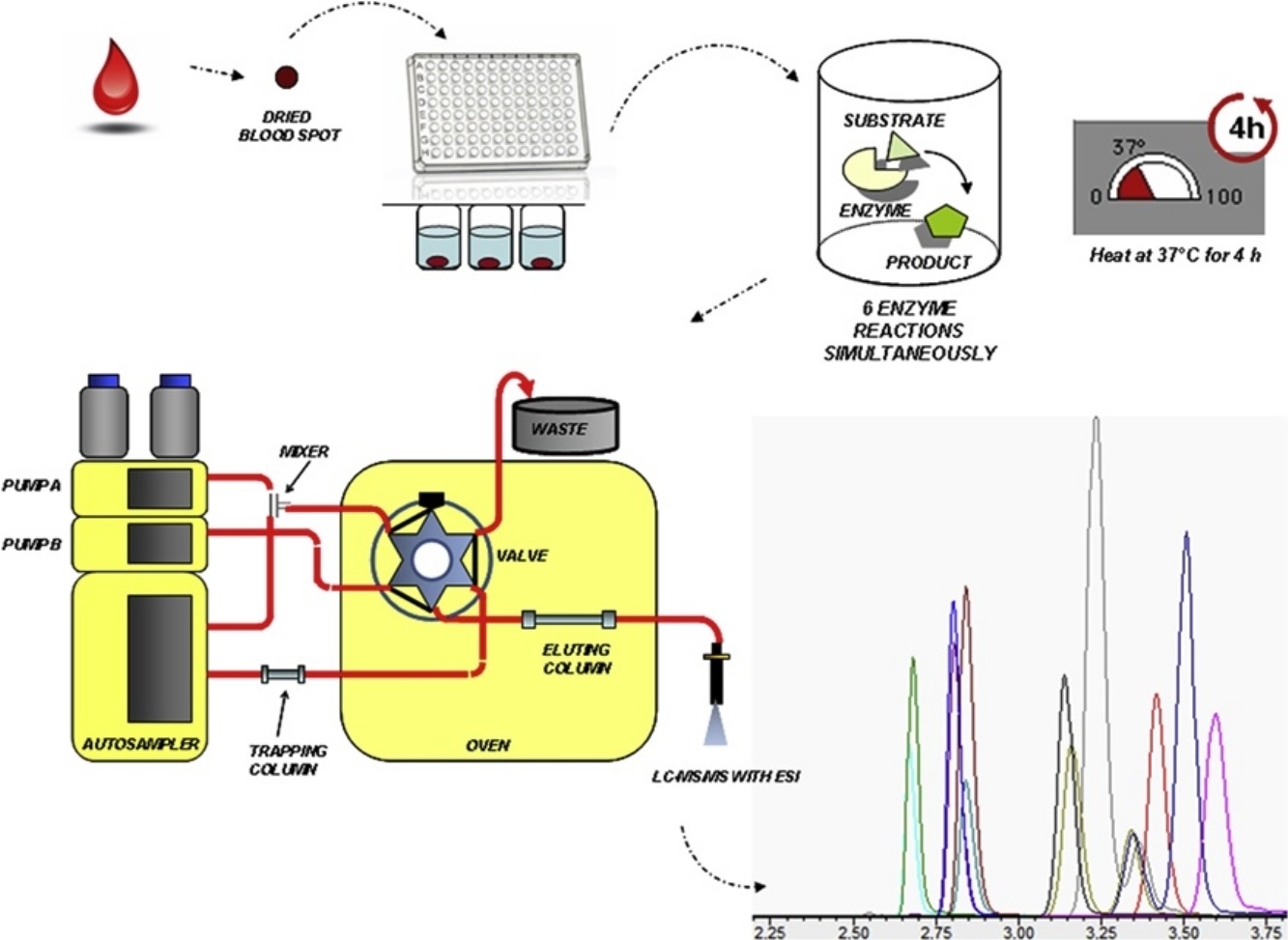 Kỹ thuật dùng Kít IVD LSDs trên LCMSMS Xác định hoạt độ 06 enzyme trong mẫu máu khô, hỗ trợ sàng lọc rối loạn dự trữ lysosome (LSD) ở trẻ sơ sinh.
|  Kỹ thuật vi lưu chất kỹ thuật số (DMF) phát hiện hoạt độ 04 Enzyme trong mẫu máu khô, hỗ trợ sàng lọc rối loạn dự trữ lysosome (LSD) ở trẻ sơ sinh
|
- Link tham khảo Phương pháp phân tích Sàng lọc rối loạn dự trữ lysosome (LSD Lysosomal Storage Disorders) kỹ thuật LCMSMS với bộ làm sạch mẫu online (Automated On-Line Sample Clean-up) trên máy SCIEX
- Lysosomal Disorders Screening in DBS LC-MS/MS Analysis Kit
1. Các nhóm bệnh rối loạn chuyển hóa bẩm sinh thường gặp ở trẻ sơ sinh:
1.1 Rối loạn chuyển hoá axit amin ở trẻ sơ sinh
- Rối loạn chuyển hóa axit amin là tình trạng các gen bị đột biến làm enzym không thể tổng hợp, phân giải hay hấp thu axit amin từ các loại thức ăn chứa protein như thịt, cá, trứng, sữa,… Điều này dẫn đến sự tích tụ các protein chưa chuyển hóa và hình thành các chất chuyển hóa bất thường trong tế bào và cơ quan, gây bệnh ở trẻ.
1.2 Rối loạn chuyển hoá axit béo ở trẻ sơ sinh
- Rối loạn chuyển hoá axit béo ở trẻ sơ sinh là một hội chứng bẩm sinh, ảnh hưởng tới khả năng phân giải mỡ ở trẻ sơ sinh. Thông thường, cơ thể sẽ chuyển hóa glucose từ tinh bột và đường để tạo ra năng lượng cho các hoạt động sống và khi nguồn năng lượng này bị cạn kiệt thì cơ thể sẽ sử dụng nguồn năng lượng dự trữ từ chất béo. Tuy nhiên, khi trẻ mắc hội chứng rối loạn chuyển hóa axit béo thì chúng sẽ không thể sử dụng được chất béo để sản sinh năng lượng. Do đó, nồng độ đường huyết của trẻ mắc bệnh luôn ở mức thấp và máu tích lũy nhiều chất độc hại.
1.3 Rối loạn chuyển hóa axit hữu cơ ở trẻ sơ sinh
- Rối loạn chuyển hóa axit hữu cơ là tình trạng rối loạn này gây ảnh hưởng đến khả năng sử dụng các amino acid (thành phần để xây dựng các protein trong cơ thể) và khả năng tổng hợp và sử dụng ketone (hợp chất hữu cơ được cơ thể tạo ra trong quá trình đốt cháy chất béo để sinh năng lượng) trong cơ thể. Khi sử dụng vitamin, protein và carbohydrate (đường) trong thực phẩm bệnh nhân có thể xuất hiện các triệu chứng nghiêm trọng, thậm chí tử vong.
- Các rối loạn chuyển hóa axit hữu cơ thường gặp gồm:
- Thiếu hụt enzyme 3-methylcrotonyl-CoA carboxylase (3MCC).
- Thiếu hụt enzyme Beta-ketothiolase (BKT).
- Bệnh Glutaric acid máu typ I (Glutaric acidemia type 1 – GA1).
- Thiếu hụt enzyme Hydroxymethyl Glutaric aciduria (HMG).
- Không thể phân giải leucin gây Kất thường acid Isovaleric (IVA).
- Bất thường trong chuyển hóa acid Methylmalonic (CBI A và CBI B).
- Thiếu hụt enzym Multiple carboxylase (MCD).
- Rối loạn chuyển hoá acid propionic (PROP).
- Trong đó, 3MCC và PROP là hai loại rối loạn thường gặp nhất, có tỷ lệ mắc là 1:75,000 trẻ sinh ra mỗi năm.
1.4 Rối loạn chuyển hoá carbonhydrat: Galactosemia
- Rối loạn chuyển hoá carbonhydrat (bệnh Galactosemia) là tình trạng dư thừa galactose trong máu. Galactose là một thành phần có trong đường lactose, thường được tìm thấy trong các sản phẩm bơ sữa, đặc biệt là trong sữa bột nhân tạo dành cho trẻ. Ngoài ra, một số thực phẩm khác cũng có thể có một lượng nhỏ galactose.
1.5 Rối loạn dự trữ thể tiêu bào (Lysosome): Pompe, MPS, Gaucher, Fabry
- Tiêu bào (Lysosome) là một bào quan chứa các enzym thuỷ phân có tác dụng làm giáng hóa protein, phức hợp carbohydrates, nucleic acid, lipids, sulfates và phosphates, tạo thành các sản phẩm có thể được tái sử dụng hoặc bị loại ra ngoài cơ thể.
- Rối loạn dự trữ thể tiêu bào là một nhóm bệnh có liên quan đến khoảng 40 rối loạn về gen mã hoá của các enzym trong lysosome. Sự không có hoặc mất chức năng của một enzyme, sau một thời gian, sẽ gây tích tụ các sản phẩm chuyển hoá trung gian, dẫn tới phá huỷ cấu trúc và chức năng tế bào. Một lượng lớn các tế bào chết đi gây ra tình trạng mất chức năng cơ quan tổ chức trong cơ thể.
- Trong đó, các bệnh rối loạn dự trữ tiêu bào Lysosome thường gặp gồm:
- Bệnh Pompe: có tỷ lệ mắc là 1:40,000.
- Bệnh MPS: bao gồm MPS-I (1:100,000), MPS-II (1:136,000), MPS-IVA (1:250,000), MPS-VI (1:300,000).
- Bệnh Gaucher: có tỷ lệ mắc là 1:57,000.
- Bệnh Fabry: có tỷ lệ mắc là 1:40,000.
2. Các phương pháp sàng lọc bệnh rối loạn dự trữ tiêu bào Lysosome với trẻ sơ sinh:
- Hiện có 02 kỹ thuật phổ biến cho sàng lock bệnh rối loạn dự trữ tiêu bào Lysosome:
- Kỹ thuật MSMS Enzymatic
- Kỹ thuật DMF Enzymatic
- So sánh 02 kỹ thuật:

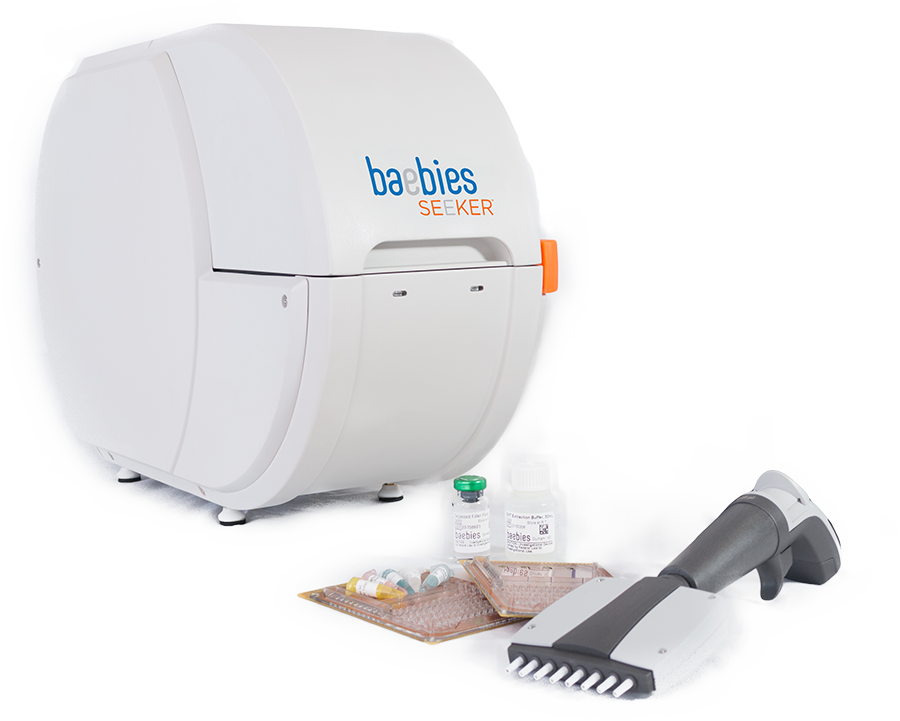
2.1 Kỹ thuật sàng lọc bệnh rối loạn dự trữ tiêu bào với kỹ thuật DMF Enzymatic
- Giải pháp hãng BABEIES – Mỹ
- Link website FDA: https://www.fda.gov/media/99718/download
- Video giới thiệu BAEBIES SEEKER Newborn Screening:
- Baebies, Inc., vào năm 2017 đã được sự chấp thuận từ Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỹ (PDA) cho sản phẩm giải pháp SEEKER. SEEKER cho đo định lượng các hoạt động của enzymes lysosomal (1) α-L-iduronidase (IDUA), (2) α-D-glucosidase (GAA), (3) β-glucocerebrosidase (GBA) (4) α-D-galactosidase A (GLA) từ mẫu giọt máu khô của trẻ sơ sinh. Giảm hoạt độ của các enzymes có thể chỉ ra các bệnh tích tụ Lysosome (LSD) Mucopolysaccharidosis Type I (MPS I), Pompe, Gaucher hoặc Fabry.
Kỹ thuật vi lưu chất kỹ thuật số (DMF) phát hiện hoạt độ Enzyme:
- Kỹ thuật vi lưu chất kỹ thuật số (DMF Digital Microfluidic Detection) sử dụng thể tích nhỏ với hệ thống đưa mẫu lỏng tự động để phân tích một lượng lớn hoạt động Enzyme sử dụng hợp chất bắt huỳnh quang (Fluorimetric substrates).
- Để phân tích hoạt độ 04 enzymes các bệnh rối loạn chuyển hoá tiêu bào LSDs,
- Quy trình phân tích:
 |
- Tương tự như phương pháp đo chuyển hoá MSMS không dẫn xuất, kỹ thuật vi lưu chất kỹ thuật số DMF phải có giải đoạn chiết mẫu trước khi đưa vào bộ cartridge DMF để phân tích. Các giọt máu khô (DBS) được chiết từ địa 96 giếng (96 well microliter plate) sẽ được đưa vào 2 tấm catridges DMF; 01 catridges để tải vào thiết bị; các bước phản ứng tiếp theo hoàn toàn tự động mà không cần bất kỳ sự can thiệp của người dùng nào. Tổng thời gian cho một chu kỳ sàng lọc xét nghiệm dùng kỹ thuật DMF cho 4 emzymes là khoảng 3,5 giờ và năng xuất đạt được 500 mẫu có thể được phân tích/ ngày.
| Enzymes | Bệnh rối loạn | |
| Bệnh Mucopolysaccharidosis I (viết tắt là MPS 1) là một dạng trong nhóm bệnh mucopolysaccharide, còn có những tên gọi khác là:
Đây là một phần trong một nhóm bệnh lớn được gọi là rối loạn dự trữ thể tiêu bào (lysosomal storage disorders – LSD) | |
| Bệnh Pompe hay bệnh dự trữ glycogen loại 2 là bệnh lý di truyền thiếu hụt enzyme acid alpha-glucosidase (GAA) trong tiêu thể khiến việc phân hủy glycogen trong tiêu thể thành glucose không thể thực hiện dẫn tới sự tích tụ glycogen trong các tế bào. Điều này làm giảm khả năng hoạt động bình thường của một số cơ quản và mô đặc biệt là cơ bắp dẫn tới các biến chứng nguy hiểm như suy tim. | |
| Bệnh Gaucher là kết quả của sự tích tụ các chất béo trong các cơ quan nhất định, đặc biệt là lá lách và gan, điều này làm cho các cơ quan trên trở nên lớn hơn nhiều so với bình thường và có thể ảnh hưởng đến chức năng của chúng. Các chất béo liên quan đến bệnh Gaucher cũng có thể tích tụ trong mô xương, gây loãng xương và tăng nguy cơ gãy xương. Nếu tủy xương bị ảnh hưởng, nó có thể gây rối loạn huyết học. | |
| Bệnh Fabry là một tình trạng di truyền hiếm gặp, khi đó cơ thể không sản xuất đủ enzyme alpha-galactosidase A (alpha-GAL). Enzyme này có vai trò phá vỡ spakenolipids, một chất giống như chất béo và ngăn chúng tích tụ trong mạch máu và mô của cơ thể. Bệnh Fabry ảnh hưởng đến tim, thận, não, hệ thần kinh trung ương và da của người bệnh. |
Quý khách có nhu cầu tư vấn, vui lòng liên hệ:
| CÔNG TY TNHH THƯƠNG MẠI – DỊCH VỤ – KỸ THUẬT VIỆT NGUYỄN | |
| Địa chỉ | VPHCM: số N36, đường số 11, P. Tân Thới Nhất, Q.12, Tp. Hồ Chí Minh. VPHN: Tòa Intracom, Số 33 Cầu Diễn, Phường Cầu Diễn, Quận Nam Từ Liêm, Hà Nội. VPĐN: Số 10 Lỗ Giáng 5, phường Hòa Xuân, quận Cẩm Lệ, Tp. Đà Nẵng. VP Cần Thơ: 275 Xuân Thủy, Phường An Bình, Quận Ninh Kiều, Tp. Cần Thơ. |
| Hotline | PHÒNG PHÁT TRIỂN SẢN PHẨM
|
| info@vietnguyenco.vn | |
| Website | https://www.vietcalib.vn| https://vietnguyenco.vn |